

2月6日,CDE发布了《化学仿制药透皮和局部给药系统黏附性和刺激性/致敏性评估临床试验技术指导原则(试行)》。对于透皮和局部给药系统仿制药,监管框架包括PK-BE、黏附性及刺激性/致敏性,兼顾体外评价与临床数据,旨在对比疗效与安全性。PK-BE可评价体内吸收速度和程度的差异,黏附性是贴剂发挥疗效的基础,直接影响药物释放的稳定性和剂量准确性,皮肤作为直接接触部位,其安全性风险需严格评估。
既往PK-BE研究与黏附性、刺激性评估常采用合并开展的简化模式。然而,随着指导原则的正式实施,行业评价标准已转向更独立、更系统、更严谨的临床研究路径。
对于黏附性评估,关键在用药部位与研究设计的适用性。若既往PK-BE试验的设计与用药部位(通常要求为“最易影响黏附性”的部位)符合新规要求,其黏附性数据可被采纳;否则,则需开展一项独立的黏附性试验,以确保评价的科学性与合规性。
对于皮肤刺激性/致敏性评估,需尤为审慎。PK-BE研究通常仅收集单次给药的刺激性评分,不足以充分反映长期或多次重复给药条件下的皮肤安全性风险。因此,开展一项独立、系统的刺激性/致敏性试验已成为满足新规要求的必要路径。
一、概念解析
接触性皮炎:指通过实验或临床观察,评估某物质(如化妆品、药品、医疗器械或化学品)在直接接触皮肤后,是否可能引发皮肤炎症反应(刺激性或过敏性)。
刺激性:物质与皮肤接触后,因化学、物理或免疫因素引起的非免疫性炎症反应,通常表现为红斑、水肿、灼痛或组织损伤,一般局限于接触部位,短期内出现和消失。
致敏性:物质通过反复接触诱导免疫系统产生过敏反应,主要为T细胞介导的迟发型超敏反应(IV型),通常可表现为丘疹、水泡、剧烈瘙痒,可扩散至非接触部位,通常延迟出现,缓慢消退。
二、评估方法及原理
1.总体设计
总体设计随机、评估者盲、受试者体内重复,可以选择参比制剂作为对照药或不含药的载体作为对照。皮肤刺激性评估应独立于先前的评估,观察者对前次评估结果保持盲态[1]。
2.研究流程
研究流程包括筛选期、21天的诱导期、14-17天的休息期、激发期(贴敷48h后观察72h)及再激发期(贴敷48h后观察72h)。对于潜在致敏性者,需要间隔4-8周后,重新进行再激发期测试[1]。诱导期用于评估多次暴露的皮肤刺激性,激发期用于评估致敏性,为实际临床使用的安全等效提供数据支持。
设计原理:
21天的反复暴露可模拟实际使用情况,观察皮肤多次暴露的刺激累积效应,并且充分激活免疫系统。参与过敏反应的朗格汉斯细胞需要多次抗原提呈以充分激活T细胞,21天可覆盖初次致敏和记忆T细胞的形成时间。
休息期(14-17天)确保T细胞分化成熟,确保记忆细胞稳定存在,并且可避免诱导期残留炎症干扰激发期结果(如假阳性)。
IV型超敏反应(T细胞介导)的峰值在接触后48-72小时。48小时接触可覆盖反应高峰,准确评估迟发型炎症。观察时长72小时,可以区别致敏性和刺激性。初次激发后间隔4-8周再次接触,可验证免疫记忆是否长期存在。皮肤表皮细胞的生长周期为28天,4-8周间隔确保皮肤免疫系统恢复基线状态。确保过敏现象仅由记忆细胞响应产生。
3.给药方案与剂量
考虑需要同时给予受试制剂和对照制剂,需要结合说明书用法用量确认给药面积(如利多卡因凝剂贴膏、双氯芬酸依泊胺贴使用1/4贴)。背部、上臂和前臂屈侧皮肤有较多树突状细胞,以背部为最佳给药部位。例如多奈哌齐透皮贴说明书用药部位包括背部、上臀部、大腿外侧,但刺激性致敏性研究选择背部给药。受试制剂和对照制剂应分布在同一解剖部位对侧位置。
4.受试者人群及例数
一般选择18-65周岁健康的男性和非孕女性,特殊品种应选择其用药人群,如雌二醇贴采用绝经期妇女进行试验。FDA指导原则要求至少200例可评价的受试者[2]。该试验周期长,脱落风险高,通常按脱落率30%计算,需入组260例左右。
● 重点排除受试者要求[1-2]
● 妊娠期或哺乳期女性;
● 有显著皮肤疾病或状况的病史,如特应性体质、银屑病、白癜风,或已知会改变皮肤外观或生理反应的情况(例如,糖尿病或卟啉病)
● 有会显著影响免疫反应的病史(例如,原发性或获得性免疫缺陷,如HIV或艾滋病;过敏性疾病,如过敏性休克、哮喘或全身性药物反应;肿瘤,如淋巴瘤或白血病;类风湿性关节炎;或系统性红斑狼疮)
● 有显著皮肤癌的病史(例如,黑色素瘤或鳞状细胞癌),浅表且未涉及透皮给药系统(TDS)应用部位的基底细胞癌除外;
● 在研究治疗开始前3周内,使用可能会:(1)显著影响或夸大对T或R产品的反应,或(2)改变对T或R产品的炎症或免疫反应的药物或治疗(例如,环孢素、他克莫司、全身或局部皮质类固醇、细胞毒性药物、免疫球蛋白、卡介苗免疫疗法、单克隆抗体或放射治疗);
● 在研究治疗开始前72小时内,使用抗组胺药或在TDS部位使用局部药物;
● 受试者两臂/左右背之间皮肤颜色存在明显差异,或存在皮肤状况,如应用部位的过多毛发、疤痕组织、纹身、开放性伤口、晒伤或身体穿孔(如纹身、穿耳洞等)可能干扰测试物品的使用、皮肤评估或受试者对透皮给药系统(TDS)的反应。
另外,可参考国内《人体皮肤斑贴试验技术指导原则(征求意见稿)》[3]中受试者的选择标准。
5.评价指标及要求[1]
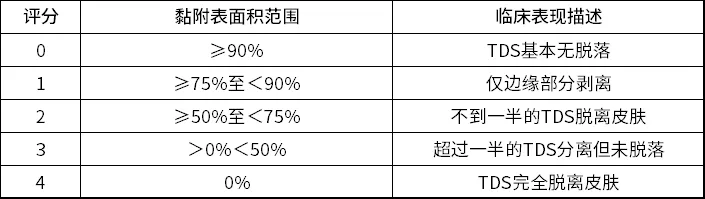
CDE推荐采用“皮肤反应”和“其他效应”评分量表进行皮肤刺激性反应评分,采用黏附性评分表进行黏附情况记录,评分量表见表1-表3。诱导期每次去除TDS后30min评价,激发期应在移除后30min、24h、48h和72h评估皮肤反应及是否致敏。
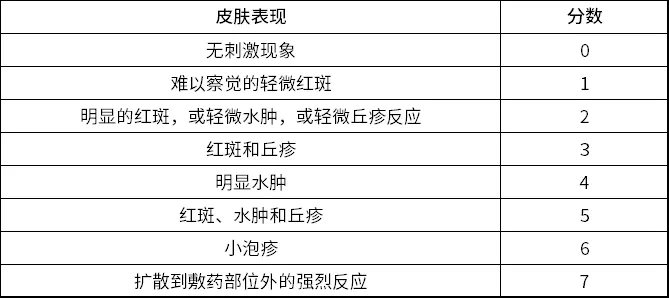
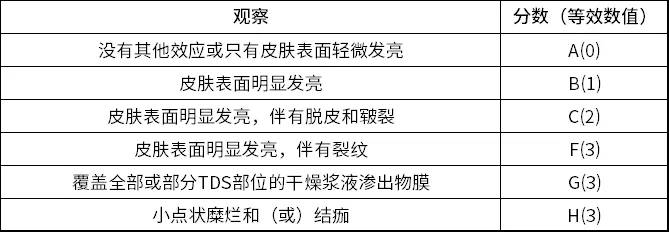
致敏性:主要评价每种TDS中观察到的可能存在潜在致敏反应的参与者数量和百分比(%),需证明仿制药与RLD TDS的致敏性相似。可能致敏的评价标准:①在激发期TDS移除后超过24小时(如48或72小时),至少完成1次评估;②最后1次激发期评估的综合刺激评分≥2;③若完成再激发测试,则在激发期和再激发期均需满足上述2个标准。另外需提供①频率表,展示激发期内每个TDS单元的敷用数量,及评价时间点的皮肤反应数字评分和其他效应字母评分;②对于所有在激发期去除TDS后48或72小时,有1个2分及以上综合刺激评分的参与者,需提供记录诱导期和激发期各时间点的实际评分的表格。
黏附性:评价数据用于监测接触充分性,确保成功诱导皮肤I/S反应,并不要求进行非劣统计,但需要在研究方案中提前设定胶带或覆盖物加固分离TDS的标准。
总结
经皮给药制剂的成分和组成,包括原料药的性质和(或)经皮给药材料阻止水蒸气从皮肤发散的程度,以及其他因素,如环境湿度或皮肤状态,可能刺激皮肤或导致致敏反应。刺激性和致敏性可能只在少数使用该产品的患者中发生,但即使这种情况发生的频率很低,不良反应也可能影响数千人。刺激性与致敏性研究对于实际临床应用具有重要意义,值得去探讨和研究。
基于对行业监管趋势的理解与前瞻性技术布局,晶易医药前期完成了多个国外审评报告、个药指导原则的调研和信息汇总,并在征求意见阶段积极反馈,其中针对不同品种的给药部位、贴敷时长等设计细节意见均被采纳,助力行业标准的完善。晶易医药不仅是新规的跟进者,更是先行实践者。
目前,公司已完成符合最新指导原则要求的皮肤刺激性及致敏性评价方法学体系的全面构建与验证,并在实际项目中积累了宝贵的成功经验。依托已验证的方法与经验,团队可提供从策略咨询、方案设计、研究执行到申报支持的全周期解决方案,助力解决透皮或局部给药项目在新旧标准衔接、临床方案设计或评价方法合规等方面的挑战,高效推进研发与注册进程。
参考文献
[1]国家药品监督管理局药品审评中心.化学仿制药透皮和局部给药系统黏附性和刺激性/致敏性评估临床试验技术指导原则(试行).2026年2月.
[2]FDA.Assessing the Irritation and Sensitization Potential of Transdermal and Topical Delivery Systems for ANDAs Guidance for Industry.April 2023 Generic Drugs Revision 1
[3]中国食品药品检定研究院.人体皮肤斑贴试验技术指导原则(征求意见稿).2023年4月
